ℹ️ Skipped - page is already crawled
| Filter | Status | Condition | Details |
|---|---|---|---|
| HTTP status | PASS | download_http_code = 200 | HTTP 200 |
| Age cutoff | PASS | download_stamp > now() - 6 MONTH | 1.3 months ago |
| History drop | PASS | isNull(history_drop_reason) | No drop reason |
| Spam/ban | PASS | fh_dont_index != 1 AND ml_spam_score = 0 | ml_spam_score=0 |
| Canonical | PASS | meta_canonical IS NULL OR = '' OR = src_unparsed | Not set |
| Property | Value |
|---|---|
| URL | https://michaelhalassa.substack.com/p/did-john-nash-really-have-schizophrenia |
| Last Crawled | 2026-03-12 01:37:04 (1 month ago) |
| First Indexed | not set |
| HTTP Status Code | 200 |
| Meta Title | Did John Nash Really Have Schizophrenia? |
| Meta Description | Many of us watched “A Beautiful Mind”, the film in which Russell Crowe played John Nash. Crowe’s performance was magnificent and I still recall the way he captured Nash’s speech upon receiving the Nobel Prize in Economic Sciences in 1994. To me, it was a story about the resilience of the human mind. |
| Meta Canonical | null |
| Boilerpipe Text | Many of us watched “
A Beautiful Mind
”, the film in which Russell Crowe played
John Nash
. Crowe’s performance was magnificent and I still recall the way he captured Nash’s speech upon receiving the Nobel Prize in Economic Sciences in 1994. To me, it was a story about the resilience of the human mind.
Fast forward to my psychiatry residency, and I am now seeing many people who carry a diagnosis of schizophrenia. I can’t see a John Nash anywhere, and the whole story began to trouble me in a different way. For the patients I see,
many had been cognitively compromised well before their first psychotic break
, and after it, some appeared to decline further. Despite adequate treatment of psychotic symptoms, a substantial portion of patients never return to their baseline, never join the workforce, and never have families of their own. This is the devastating picture that dominates clinical training, and it sits uneasily alongside the story of a man who spent years still doing mathematics at Princeton after his psychosis had largely remitted. At some point I began to wonder: did John Nash really have schizophrenia?
The Nash question is in some ways unanswerable, and I have made my peace with that.
In a previous piece
, I argued that schizophrenia is almost certainly a collection of many entities, and that our diagnostic labels, schizophrenia, schizoaffective disorder, bipolar disorder with psychotic features, carve nature at joints that may not exist or be the best guides for treatment. These categories describe collections of behaviors that may not map on distinct mechanisms or generative processes. From that vantage point, asking whether Nash had “real” schizophrenia or bipolar disorder may be ill posed. The labels are placeholders for something we do not yet understand.
And yet the question refuses to go away, because the clinical differences are real even if the categories are artificial. It is true that I never saw something as dramatic as a beautiful mind, but some people with a schizophrenia diagnosis can have relatively intact cognitive capacity. These people are quite different from those whose cognition appears to deteriorate, akin to Kraepelin’s
dementia praecox
description. Something is producing that difference, even if our nosology cannot name it.
A paper just published in Molecular Psychiatry by Watson and colleagues
takes a step toward naming it, using genetics to do something that clinical observation alone has never managed: begin to show us what is actually varying underneath the label.
The previous piece I wrote on schizophrenia
argued that the heterogeneity we observe clinically may reflect distinct generative processes rather than variation of a singular category. In other words, among the vast diversity within the
psychosis spectrum
, we can appreciate at least two distinct trajectories: patients who arrive at their first psychotic break
already cognitively compromised
, and who face a ceiling on recovery that most cannot exceed, and those whose cognition remains relatively intact, but with the reasoning changes that define psychotic illnesses more generally. If these were simply different severities of one process, you would expect them to respond similarly to treatment and follow predictable paths. They do not.
The question is what to do with that observation. Current clinical tools cannot resolve it. The acute presentation looks the same across both trajectories, and the longitudinal picture only becomes clear years later, often after multiple diagnostic revisions and treatment trials that may not have been appropriate for either. What we need is a way to probe the biology that is upstream of symptoms and independent of the categorical labels that symptoms generate. That is a harder problem than it sounds, and genetics is only one partial answer to it.
Heritability estimates for schizophrenia hover around
~64%
(but removing shared environment, including that in the womb, reduces that further), so genetics alone cannot be the answer and the gap between genetic markers and any clinically actionable prediction remains vast. What genetics appears to be able to do, more modestly, is act as a probe for mechanistic coherence. If a diagnostic category is compressing patients with genuinely distinct underlying biology, the genetic signal inside that category should reflect the compression. It should look composite, pulling in different directions depending on which patients you happen to be sampling. And if you can decompose that signal, you can start to ask whether the clinical heterogeneity you observe maps onto something with a biological basis, even if you cannot yet name the mechanisms.
The cancer parallel is instructive here, even if imperfect. In chronic myelogenous leukemia, a cancer of the blood and bone marrow, decades of clinical heterogeneity (why some patients deteriorated faster, why responses varied) turned out to reflect, in a substantial proportion of cases, the presence of the Philadelphia chromosome. This is a translocation, or swap of genetic material, between chromosomes 9 and 22 that generates an abnormal fusion gene and drives uncontrolled cell growth. Once that mechanism was identified, a drug could be designed against it specifically. Imatinib,
approved in 2001
, transformed what had been a death sentence into a manageable condition for many patients. What is critical to understand here is that decomposing a clinically heterogeneous category by its underlying biology changed what was therapeutically possible. Whether that kind of decomposition is achievable in psychosis remains an open question, but Watson and colleagues take a step in that direction, demonstrating that the genetic signal underneath a single diagnostic label is not unitary.
Beginning in the late 2000s, genome-wide association studies (GWAS) made it possible to scan hundreds of thousands of genetic variants across large populations simultaneously, searching for common variants that each contribute a small increment of disease risk. For schizophrenia, the effort eventually identified hundreds of genomic loci. But rather than converging on a coherent biological picture, the signal appeared to pull in multiple directions at once, and understanding why took the better part of a decade.
The first hint of a composite signal came from an apparent paradox within the data itself. At the phenotypic level, schizophrenia is associated with lower educational attainment and cognitive decline. You would therefore expect the genetic risk architecture to show a negative correlation with both. It does not. The genetic correlation between schizophrenia risk and IQ is negative, as expected, but the correlation with educational attainment is weakly positive, meaning the same variants that increase schizophrenia risk also associate, at the population level, with staying in school longer. This counterintuitive finding was reported across multiple large studies and resisted easy explanation.
The resolution, proposed by
Lencz and colleagues
and subsequently replicated and extended, was that the aggregate GWAS signal was masking two biologically distinct subsets of variants. One subset showed the expected pattern: greater schizophrenia risk alongside lower cognition and lower educational attainment. These variants, when subjected to pathway analysis, pointed toward early neurodevelopmental mechanisms (genes related to how the brain was assembled during early development). A second subset showed the opposite: greater schizophrenia risk alongside higher educational attainment, and pointing towards mechanisms of synaptic function later on in life (mechanisms governing learning after birth). These two components were canceling each other out, resulting in the overall signal being near zero for educational attainment. This was an important conceptual advance, but the methods used could not formally separate and quantify the two components as independent.
A second line of evidence arrived from a different methodology entirely and pointed in a consistent direction.
Rare variant studies
, which examine low-frequency mutations with large individual effects, found that copy number variants and rare disruptive coding mutations associated with schizophrenia, at loci like 22q11.2, 1q21.1, and 15q13.3, are also enriched in autism and intellectual disability. These are high-penetrance risk factors for neurodevelopmental disruption that happen to increase the probability of psychosis among many other outcomes. Patients carrying heavier rare-variant burden tend to present with
more cognitive compromise
.
What the field lacked was a method capable of formally separating the two components of the common-variant signal into distinct genetic constructs, quantifying their independent relationships to cognition, and doing so in a framework clean enough to support downstream analyses like
polygenic scoring
and
Mendelian randomization
. That is the gap Watson and colleagues set out to close.
Watson and colleagues took this unresolved picture and asked whether a newer statistical framework could do what prior approaches could not: treat the two components of the schizophrenia signal not as overlapping subsets but as formally independent genetic constructs. The approach they used, GWAS-by-subtraction implemented within a Genomic Structural Equation Modeling (SEM) framework, is worth understanding.
Genomic SEM works similarly in spirit to factor analysis: it models latent genetic dimensions across multiple GWAS datasets simultaneously. The specific application here is intuitive once explained. You start with the full genome-wide association signal for schizophrenia, which captures all common variants associated with the disorder. You then use the bipolar disorder GWAS signal as a genetic filter, statistically removing the variance that schizophrenia shares with affective psychosis. What remains after subtraction is the schizophrenia-specific component: genetic signal that predicts schizophrenia but is independent of affective psychosis risk. The complementary component, the shared portion, is what they call PSYshared.
The paper then asks three questions of these components: how do they correlate genetically with IQ and educational attainment (EA), do polygenic scores built from them predict educational outcomes in a large population sample, and does Mendelian randomization support a causal interpretation?
The genetic correlations are where the story sharpens. When you look at the overall schizophrenia signal, it appears to show almost no relationship with educational attainment. That turns out to be because two components are pulling in opposite directions and canceling each other out. The schizophrenia-specific component correlates negatively with both IQ and educational attainment, while the shared component correlates positively with educational attainment.
Polygenic scores built from each component and tested in nearly 382,000 people in the UK Biobank told the same story: people carrying more of the schizophrenia-specific component tended to have lower educational attainment, while those carrying more of the shared component tended to have higher educational attainment. Crucially, this was an entirely independent sample, meaning the opposing signals generalize beyond the original analysis.
Genetic correlations tell you that two things travel together in the genome, but not which way the arrow of causation points, or whether a third factor is driving both. Mendelian randomization addresses this by using genetic variants as a natural experiment. Because you inherit your genes at conception, before any life experiences accumulate, your genetic makeup cannot be caused by your educational outcomes. Watson and colleagues used this approach and found that genetic liability for the schizophrenia-specific component appears to drive lower cognition, while liability for the shared component appears to drive higher educational attainment. The caveat is that some variants may affect both psychosis risk and cognition through completely separate biological routes, so it is difficult to be certain the causal effect is running specifically through psychosis risk rather than through some other property of the same variants. That said, the Mendelian randomization results are broadly consistent with a causal relationship whose veracity will be tested in future experiments.
The final piece of the paper maps where in the brain these two genetic components are most active, using publicly available brain expression databases to ask which regions show the highest expression of the relevant genes. The shared component is predominantly expressed in cortical regions, particularly frontal cortex, consistent with the postnatally expressed synaptic biology that has been associated with affective psychosis in independent work. The schizophrenia-specific component shows a broader pattern, with expression extending into subcortical regions. This pattern is broadly consistent with the picture that has been building across the paper: the schizophrenia-specific component is not simply a more severe version of the shared one, but something with potentially distinct neural involvement. These analyses are exploratory, and the authors present them as such, but they add anatomical texture to the genetic analyses.
So where does this all leave us? Watson and colleagues provide important information about biological heterogeneity within the categorical label of schizophrenia. Maybe more importantly, they clarify the relationship between this category and two others, the psychosis spectrum and neurodevelopmental disorders.
One thing worth noting about the study is the notion of a schizophrenia-specific component. Methodologically, this component is the residual genetic signal that remains after removing shared variance with the broader psychosis spectrum. Personally, I would have probably just called it that, a residual, and not schizophrenia-specific. Because what really is schizophrenia? The neurodevelopmental framing is plausible and supported by converging evidence, but the label risks conferring more categorical definition than the method warrants.
Over many previous pieces in this newsletter, I have made a prediction that what we call schizophrenia will eventually give way to a set of subtypes defined by computational processes altered in individual people. These are algorithmic-level changes that ultimately impact how people build models of the world, update them, and approach reasoning challenges more generally. Two patients carrying the same diagnosis may have distinct algorithmic-level changes that would be described similarly by natural language, the substrate of clinical communication. Do genetics help with this computational dissection? Not directly, but they would add some confidence to conclusions drawn at this downstream process-level description. For grand challenges like schizophrenia, we will need multiple different methods to triangulate the generative process. Genetics underlie vulnerabilities, akin to the hyperparameters that set up model architectures in AI, but all the training data, inputs through early development and later fine-tuning via life, will determine the algorithmic-level changes that shape clinical presentation, and perhaps even treatment response.
So did John Nash really have schizophrenia? The film that brought his story to millions took considerable liberties with the clinical record: his hallucinations were auditory, not the elaborate visual conspiracies Hollywood rendered; and his greatest mathematical work was complete before psychosis ever emerged. There had been
some discussion
regarding whether his condition was truly schizophrenia or bipolar, but that debate may miss the point entirely. What's clear from the record is that Nash was not a man who did mathematics through schizophrenia, he was someone whose psychosis arrived after his most important work was done, perhaps ran an episodic illness and eventually receded, leaving behind someone who was diminished in some ways but still capable of serious thought. That trajectory does not sit outside the distribution of what we call schizophrenia.
If individual people carry the neurodevelopmental and affective psychosis components in continuously varying proportions, then Nash's case stops being a paradox and becomes exactly what we should expect to find somewhere in that space.
His case has been sitting at the edge of our nosology for decades, neither fully explained nor fully dismissed. Now we have tools to reframe the question entirely.
If you found this piece useful, please consider subscribing and sharing it with someone who might appreciate it too.
Acknowledgement: I am grateful for Dr.
Cameron Watson
’s input on this post. Please check him out and follow his work. I know I will!
Cameron is a Specialist Registrar in Psychiatry at the Maudsley Hospital in London and a UKRI Medical Research Council Clinical Research Training Fellow at the Institute of Psychiatry, Psychology and Neuroscience, King’s College London. His research integrates cognitive, genetic, and neurophysiological data to disentangle heterogeneity within psychotic disorders, with the ultimate aim of improving understanding of the diverse mechanisms driving these conditions.
No posts |
| Markdown | [](https://michaelhalassa.substack.com/)
# [](https://michaelhalassa.substack.com/)
Subscribe
Sign in
# Did John Nash Really Have Schizophrenia?
[](https://substack.com/@michaelhalassa)
[Michael Halassa](https://substack.com/@michaelhalassa)
Mar 07, 2026
48
33
17
Share
Many of us watched “[A Beautiful Mind](https://en.wikipedia.org/wiki/A_Beautiful_Mind_\(film\))”, the film in which Russell Crowe played [John Nash](https://en.wikipedia.org/wiki/John_Forbes_Nash_Jr.). Crowe’s performance was magnificent and I still recall the way he captured Nash’s speech upon receiving the Nobel Prize in Economic Sciences in 1994. To me, it was a story about the resilience of the human mind.
[](https://substackcdn.com/image/fetch/$s_!AoqN!,f_auto,q_auto:good,fl_progressive:steep/https%3A%2F%2Fsubstack-post-media.s3.amazonaws.com%2Fpublic%2Fimages%2F616133bf-0f98-4eeb-8626-b93793baddbf_744x496.png)
Fast forward to my psychiatry residency, and I am now seeing many people who carry a diagnosis of schizophrenia. I can’t see a John Nash anywhere, and the whole story began to trouble me in a different way. For the patients I see, [many had been cognitively compromised well before their first psychotic break](https://psychiatryonline.org/doi/10.1176/appi.ajp.2009.09040574), and after it, some appeared to decline further. Despite adequate treatment of psychotic symptoms, a substantial portion of patients never return to their baseline, never join the workforce, and never have families of their own. This is the devastating picture that dominates clinical training, and it sits uneasily alongside the story of a man who spent years still doing mathematics at Princeton after his psychosis had largely remitted. At some point I began to wonder: did John Nash really have schizophrenia?
The Nash question is in some ways unanswerable, and I have made my peace with that. [In a previous piece](https://michaelhalassa.substack.com/p/what-will-schizophrenia-become), I argued that schizophrenia is almost certainly a collection of many entities, and that our diagnostic labels, schizophrenia, schizoaffective disorder, bipolar disorder with psychotic features, carve nature at joints that may not exist or be the best guides for treatment. These categories describe collections of behaviors that may not map on distinct mechanisms or generative processes. From that vantage point, asking whether Nash had “real” schizophrenia or bipolar disorder may be ill posed. The labels are placeholders for something we do not yet understand.
And yet the question refuses to go away, because the clinical differences are real even if the categories are artificial. It is true that I never saw something as dramatic as a beautiful mind, but some people with a schizophrenia diagnosis can have relatively intact cognitive capacity. These people are quite different from those whose cognition appears to deteriorate, akin to Kraepelin’s [dementia praecox](https://en.wikipedia.org/wiki/Dementia_praecox) description. Something is producing that difference, even if our nosology cannot name it. [A paper just published in Molecular Psychiatry by Watson and colleagues](https://www.nature.com/articles/s41380-026-03444-3) takes a step toward naming it, using genetics to do something that clinical observation alone has never managed: begin to show us what is actually varying underneath the label.
***
## **Two Trajectories Inside One Label**
[The previous piece I wrote on schizophrenia](https://michaelhalassa.substack.com/p/what-will-schizophrenia-become) argued that the heterogeneity we observe clinically may reflect distinct generative processes rather than variation of a singular category. In other words, among the vast diversity within the [psychosis spectrum](https://pmc.ncbi.nlm.nih.gov/articles/PMC11615062/), we can appreciate at least two distinct trajectories: patients who arrive at their first psychotic break [already cognitively compromised](https://www.nature.com/articles/s41398-023-02718-6), and who face a ceiling on recovery that most cannot exceed, and those whose cognition remains relatively intact, but with the reasoning changes that define psychotic illnesses more generally. If these were simply different severities of one process, you would expect them to respond similarly to treatment and follow predictable paths. They do not.
The question is what to do with that observation. Current clinical tools cannot resolve it. The acute presentation looks the same across both trajectories, and the longitudinal picture only becomes clear years later, often after multiple diagnostic revisions and treatment trials that may not have been appropriate for either. What we need is a way to probe the biology that is upstream of symptoms and independent of the categorical labels that symptoms generate. That is a harder problem than it sounds, and genetics is only one partial answer to it.
Heritability estimates for schizophrenia hover around [~64%](https://pubmed.ncbi.nlm.nih.gov/19150704/) (but removing shared environment, including that in the womb, reduces that further), so genetics alone cannot be the answer and the gap between genetic markers and any clinically actionable prediction remains vast. What genetics appears to be able to do, more modestly, is act as a probe for mechanistic coherence. If a diagnostic category is compressing patients with genuinely distinct underlying biology, the genetic signal inside that category should reflect the compression. It should look composite, pulling in different directions depending on which patients you happen to be sampling. And if you can decompose that signal, you can start to ask whether the clinical heterogeneity you observe maps onto something with a biological basis, even if you cannot yet name the mechanisms.
The cancer parallel is instructive here, even if imperfect. In chronic myelogenous leukemia, a cancer of the blood and bone marrow, decades of clinical heterogeneity (why some patients deteriorated faster, why responses varied) turned out to reflect, in a substantial proportion of cases, the presence of the Philadelphia chromosome. This is a translocation, or swap of genetic material, between chromosomes 9 and 22 that generates an abnormal fusion gene and drives uncontrolled cell growth. Once that mechanism was identified, a drug could be designed against it specifically. Imatinib, [approved in 2001](https://www.nejm.org/doi/full/10.1056/NEJM200104053441401), transformed what had been a death sentence into a manageable condition for many patients. What is critical to understand here is that decomposing a clinically heterogeneous category by its underlying biology changed what was therapeutically possible. Whether that kind of decomposition is achievable in psychosis remains an open question, but Watson and colleagues take a step in that direction, demonstrating that the genetic signal underneath a single diagnostic label is not unitary.
***
## **What the Genetic Signal Inside “Schizophrenia” Actually Looks Like**
Beginning in the late 2000s, genome-wide association studies (GWAS) made it possible to scan hundreds of thousands of genetic variants across large populations simultaneously, searching for common variants that each contribute a small increment of disease risk. For schizophrenia, the effort eventually identified hundreds of genomic loci. But rather than converging on a coherent biological picture, the signal appeared to pull in multiple directions at once, and understanding why took the better part of a decade.
The first hint of a composite signal came from an apparent paradox within the data itself. At the phenotypic level, schizophrenia is associated with lower educational attainment and cognitive decline. You would therefore expect the genetic risk architecture to show a negative correlation with both. It does not. The genetic correlation between schizophrenia risk and IQ is negative, as expected, but the correlation with educational attainment is weakly positive, meaning the same variants that increase schizophrenia risk also associate, at the population level, with staying in school longer. This counterintuitive finding was reported across multiple large studies and resisted easy explanation.
The resolution, proposed by [Lencz and colleagues](https://www.cell.com/ajhg/fulltext/S0002-9297\(19\)30237-X?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS000292971930237X%3Fshowall%3Dtrue) and subsequently replicated and extended, was that the aggregate GWAS signal was masking two biologically distinct subsets of variants. One subset showed the expected pattern: greater schizophrenia risk alongside lower cognition and lower educational attainment. These variants, when subjected to pathway analysis, pointed toward early neurodevelopmental mechanisms (genes related to how the brain was assembled during early development). A second subset showed the opposite: greater schizophrenia risk alongside higher educational attainment, and pointing towards mechanisms of synaptic function later on in life (mechanisms governing learning after birth). These two components were canceling each other out, resulting in the overall signal being near zero for educational attainment. This was an important conceptual advance, but the methods used could not formally separate and quantify the two components as independent.
A second line of evidence arrived from a different methodology entirely and pointed in a consistent direction. [Rare variant studies](https://www.nature.com/articles/nature07229), which examine low-frequency mutations with large individual effects, found that copy number variants and rare disruptive coding mutations associated with schizophrenia, at loci like 22q11.2, 1q21.1, and 15q13.3, are also enriched in autism and intellectual disability. These are high-penetrance risk factors for neurodevelopmental disruption that happen to increase the probability of psychosis among many other outcomes. Patients carrying heavier rare-variant burden tend to present with [more cognitive compromise](https://www.biologicalpsychiatryjournal.com/article/S0006-3223\(20\)32117-X/abstract).
What the field lacked was a method capable of formally separating the two components of the common-variant signal into distinct genetic constructs, quantifying their independent relationships to cognition, and doing so in a framework clean enough to support downstream analyses like [polygenic scoring](https://www.genome.gov/Health/Genomics-and-Medicine/Polygenic-risk-scores) and [Mendelian randomization](https://en.wikipedia.org/wiki/Mendelian_randomization). That is the gap Watson and colleagues set out to close.
***
## **What Watson and Colleagues Add**
Watson and colleagues took this unresolved picture and asked whether a newer statistical framework could do what prior approaches could not: treat the two components of the schizophrenia signal not as overlapping subsets but as formally independent genetic constructs. The approach they used, GWAS-by-subtraction implemented within a Genomic Structural Equation Modeling (SEM) framework, is worth understanding.
Genomic SEM works similarly in spirit to factor analysis: it models latent genetic dimensions across multiple GWAS datasets simultaneously. The specific application here is intuitive once explained. You start with the full genome-wide association signal for schizophrenia, which captures all common variants associated with the disorder. You then use the bipolar disorder GWAS signal as a genetic filter, statistically removing the variance that schizophrenia shares with affective psychosis. What remains after subtraction is the schizophrenia-specific component: genetic signal that predicts schizophrenia but is independent of affective psychosis risk. The complementary component, the shared portion, is what they call PSYshared.
[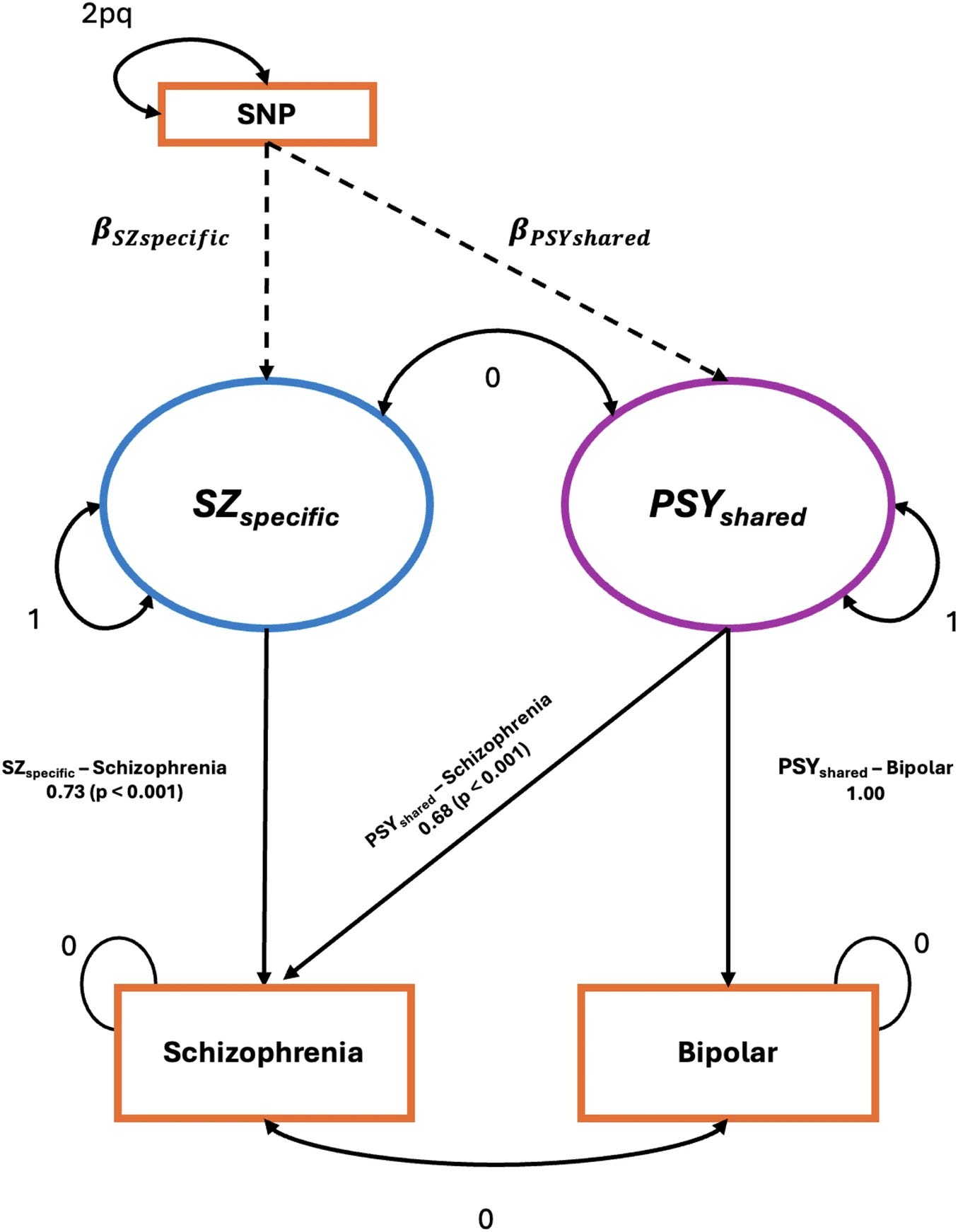](https://substackcdn.com/image/fetch/$s_!7uCD!,f_auto,q_auto:good,fl_progressive:steep/https%3A%2F%2Fsubstack-post-media.s3.amazonaws.com%2Fpublic%2Fimages%2Ff6490a4c-e063-4c5d-a324-015d17e35028_1355x1742.png)
The paper then asks three questions of these components: how do they correlate genetically with IQ and educational attainment (EA), do polygenic scores built from them predict educational outcomes in a large population sample, and does Mendelian randomization support a causal interpretation?
The genetic correlations are where the story sharpens. When you look at the overall schizophrenia signal, it appears to show almost no relationship with educational attainment. That turns out to be because two components are pulling in opposite directions and canceling each other out. The schizophrenia-specific component correlates negatively with both IQ and educational attainment, while the shared component correlates positively with educational attainment.
[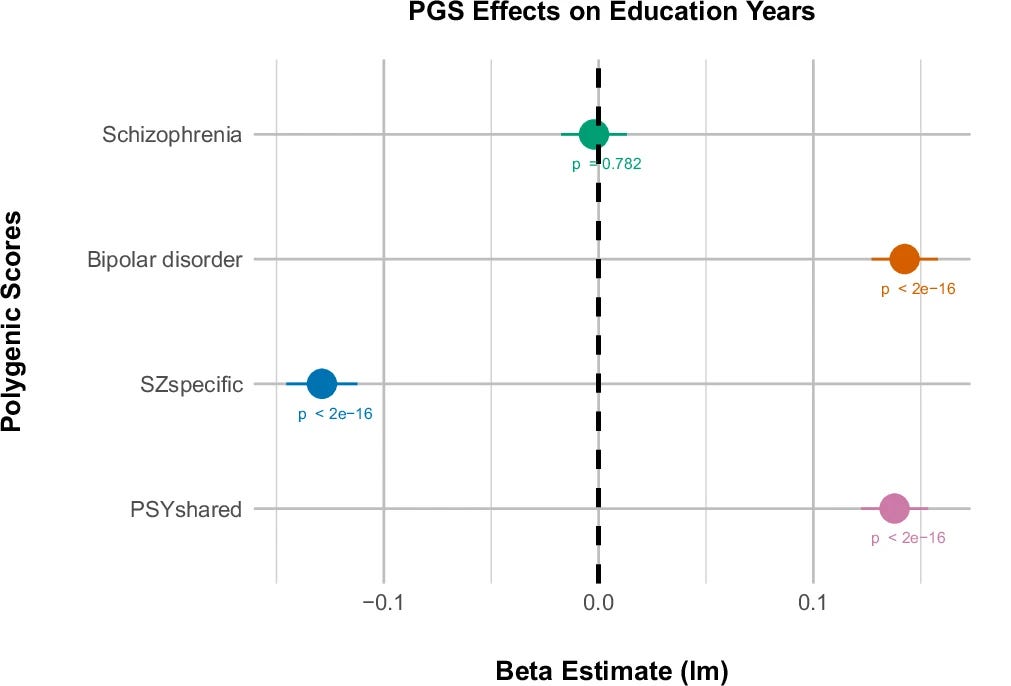](https://substackcdn.com/image/fetch/$s_!tnah!,f_auto,q_auto:good,fl_progressive:steep/https%3A%2F%2Fsubstack-post-media.s3.amazonaws.com%2Fpublic%2Fimages%2Facff7772-5eda-4a9c-b5fa-c1a06eda6af1_1011x686.png)
Polygenic scores built from each component and tested in nearly 382,000 people in the UK Biobank told the same story: people carrying more of the schizophrenia-specific component tended to have lower educational attainment, while those carrying more of the shared component tended to have higher educational attainment. Crucially, this was an entirely independent sample, meaning the opposing signals generalize beyond the original analysis.
Genetic correlations tell you that two things travel together in the genome, but not which way the arrow of causation points, or whether a third factor is driving both. Mendelian randomization addresses this by using genetic variants as a natural experiment. Because you inherit your genes at conception, before any life experiences accumulate, your genetic makeup cannot be caused by your educational outcomes. Watson and colleagues used this approach and found that genetic liability for the schizophrenia-specific component appears to drive lower cognition, while liability for the shared component appears to drive higher educational attainment. The caveat is that some variants may affect both psychosis risk and cognition through completely separate biological routes, so it is difficult to be certain the causal effect is running specifically through psychosis risk rather than through some other property of the same variants. That said, the Mendelian randomization results are broadly consistent with a causal relationship whose veracity will be tested in future experiments.
The final piece of the paper maps where in the brain these two genetic components are most active, using publicly available brain expression databases to ask which regions show the highest expression of the relevant genes. The shared component is predominantly expressed in cortical regions, particularly frontal cortex, consistent with the postnatally expressed synaptic biology that has been associated with affective psychosis in independent work. The schizophrenia-specific component shows a broader pattern, with expression extending into subcortical regions. This pattern is broadly consistent with the picture that has been building across the paper: the schizophrenia-specific component is not simply a more severe version of the shared one, but something with potentially distinct neural involvement. These analyses are exploratory, and the authors present them as such, but they add anatomical texture to the genetic analyses.
***
## **The Lesson and Nash**
So where does this all leave us? Watson and colleagues provide important information about biological heterogeneity within the categorical label of schizophrenia. Maybe more importantly, they clarify the relationship between this category and two others, the psychosis spectrum and neurodevelopmental disorders.
One thing worth noting about the study is the notion of a schizophrenia-specific component. Methodologically, this component is the residual genetic signal that remains after removing shared variance with the broader psychosis spectrum. Personally, I would have probably just called it that, a residual, and not schizophrenia-specific. Because what really is schizophrenia? The neurodevelopmental framing is plausible and supported by converging evidence, but the label risks conferring more categorical definition than the method warrants.
Over many previous pieces in this newsletter, I have made a prediction that what we call schizophrenia will eventually give way to a set of subtypes defined by computational processes altered in individual people. These are algorithmic-level changes that ultimately impact how people build models of the world, update them, and approach reasoning challenges more generally. Two patients carrying the same diagnosis may have distinct algorithmic-level changes that would be described similarly by natural language, the substrate of clinical communication. Do genetics help with this computational dissection? Not directly, but they would add some confidence to conclusions drawn at this downstream process-level description. For grand challenges like schizophrenia, we will need multiple different methods to triangulate the generative process. Genetics underlie vulnerabilities, akin to the hyperparameters that set up model architectures in AI, but all the training data, inputs through early development and later fine-tuning via life, will determine the algorithmic-level changes that shape clinical presentation, and perhaps even treatment response.
So did John Nash really have schizophrenia? The film that brought his story to millions took considerable liberties with the clinical record: his hallucinations were auditory, not the elaborate visual conspiracies Hollywood rendered; and his greatest mathematical work was complete before psychosis ever emerged. There had been [some discussion](https://onlinelibrary.wiley.com/doi/10.1111/acps.12504) regarding whether his condition was truly schizophrenia or bipolar, but that debate may miss the point entirely. What's clear from the record is that Nash was not a man who did mathematics through schizophrenia, he was someone whose psychosis arrived after his most important work was done, perhaps ran an episodic illness and eventually receded, leaving behind someone who was diminished in some ways but still capable of serious thought. That trajectory does not sit outside the distribution of what we call schizophrenia. **If individual people carry the neurodevelopmental and affective psychosis components in continuously varying proportions, then Nash's case stops being a paradox and becomes exactly what we should expect to find somewhere in that space.** His case has been sitting at the edge of our nosology for decades, neither fully explained nor fully dismissed. Now we have tools to reframe the question entirely.
[](https://substackcdn.com/image/fetch/$s_!PQfr!,f_auto,q_auto:good,fl_progressive:steep/https%3A%2F%2Fsubstack-post-media.s3.amazonaws.com%2Fpublic%2Fimages%2F881341fc-fc7c-4c5f-a7fc-bc73d4cda327_640x524.gif)
***
***
*If you found this piece useful, please consider subscribing and sharing it with someone who might appreciate it too.*
***
Acknowledgement: I am grateful for Dr. [Cameron Watson](https://www.kcl.ac.uk/people/cameron-watson)’s input on this post. Please check him out and follow his work. I know I will\!
[](https://substackcdn.com/image/fetch/$s_!29CN!,f_auto,q_auto:good,fl_progressive:steep/https%3A%2F%2Fsubstack-post-media.s3.amazonaws.com%2Fpublic%2Fimages%2F684d07dd-50b9-4715-b5b8-cc9f6d1db584_200x200.png)
Cameron is a Specialist Registrar in Psychiatry at the Maudsley Hospital in London and a UKRI Medical Research Council Clinical Research Training Fellow at the Institute of Psychiatry, Psychology and Neuroscience, King’s College London. His research integrates cognitive, genetic, and neurophysiological data to disentangle heterogeneity within psychotic disorders, with the ultimate aim of improving understanding of the diverse mechanisms driving these conditions.
48
33
17
Share
#### Discussion about this post
Comments
Restacks
[](https://substack.com/profile/147604702-m-stankovich-md-msw?utm_source=comment)
[M. Stankovich, MD, MSW](https://substack.com/profile/147604702-m-stankovich-md-msw?utm_source=substack-feed-item)
[3d](https://michaelhalassa.substack.com/p/did-john-nash-really-have-schizophrenia/comment/225107850 "Mar 9, 2026, 4:31 AM")Edited
Liked by Michael Halassa
I agree completely with your conclusion regarding Nash. I had two highly educated patients one -an advanced healthcare provider and the second a legal professional- and they both reported frequent auditory hallucinations; episodic antipsychotic medications that they stopped because of side-effects; and they both came to me for other concerns and reported the hallucinations as part of their history. They did not, as you say, “sit outside the distribution of what we call schizophrenia.” Yet, very little about their actual life - family, home, longterm employment, etc. - appeared at odds with cognitive reality. Nevertheless, both individuals reported hearing terrifying, deprecating, sometimes instructive/commanding voices that were shocking. They certainly were instructive. A very fine article, Michael\!
[Reply]()
[Share]()
[1 reply by Michael Halassa](https://michaelhalassa.substack.com/p/did-john-nash-really-have-schizophrenia/comment/225107850)
[](https://substack.com/profile/87025564-michal-patarak?utm_source=comment)
[Michal Patarák](https://substack.com/profile/87025564-michal-patarak?utm_source=substack-feed-item)
[2d](https://michaelhalassa.substack.com/p/did-john-nash-really-have-schizophrenia/comment/225692099 "Mar 10, 2026, 12:03 PM")
Liked by Michael Halassa
In my experience, however, schizophrenia is very often diagnosed in people with autism spectrum disorder who develop psychotic symptoms. Their cognitive and personality specificities are then retrospectively reinterpreted as neurodevelopmental difficulties preceding the onset of schizophrenia.
[Reply]()
[Share]()
[2 replies by Michael Halassa and others](https://michaelhalassa.substack.com/p/did-john-nash-really-have-schizophrenia/comment/225692099)
[31 more comments...](https://michaelhalassa.substack.com/p/did-john-nash-really-have-schizophrenia/comments)
Top
Latest
Discussions
No posts
### Ready for more?
© 2026 Michael Halassa · [Privacy](https://substack.com/privacy) ∙ [Terms](https://substack.com/tos) ∙ [Collection notice](https://substack.com/ccpa#personal-data-collected)
[Start your Substack](https://substack.com/signup?utm_source=substack&utm_medium=web&utm_content=footer)
[Get the app](https://substack.com/app/app-store-redirect?utm_campaign=app-marketing&utm_content=web-footer-button)
[Substack](https://substack.com/) is the home for great culture
This site requires JavaScript to run correctly. Please [turn on JavaScript](https://enable-javascript.com/) or unblock scripts |
| Readable Markdown | Many of us watched “[A Beautiful Mind](https://en.wikipedia.org/wiki/A_Beautiful_Mind_\(film\))”, the film in which Russell Crowe played [John Nash](https://en.wikipedia.org/wiki/John_Forbes_Nash_Jr.). Crowe’s performance was magnificent and I still recall the way he captured Nash’s speech upon receiving the Nobel Prize in Economic Sciences in 1994. To me, it was a story about the resilience of the human mind.
[](https://substackcdn.com/image/fetch/$s_!AoqN!,f_auto,q_auto:good,fl_progressive:steep/https%3A%2F%2Fsubstack-post-media.s3.amazonaws.com%2Fpublic%2Fimages%2F616133bf-0f98-4eeb-8626-b93793baddbf_744x496.png)
Fast forward to my psychiatry residency, and I am now seeing many people who carry a diagnosis of schizophrenia. I can’t see a John Nash anywhere, and the whole story began to trouble me in a different way. For the patients I see, [many had been cognitively compromised well before their first psychotic break](https://psychiatryonline.org/doi/10.1176/appi.ajp.2009.09040574), and after it, some appeared to decline further. Despite adequate treatment of psychotic symptoms, a substantial portion of patients never return to their baseline, never join the workforce, and never have families of their own. This is the devastating picture that dominates clinical training, and it sits uneasily alongside the story of a man who spent years still doing mathematics at Princeton after his psychosis had largely remitted. At some point I began to wonder: did John Nash really have schizophrenia?
The Nash question is in some ways unanswerable, and I have made my peace with that. [In a previous piece](https://michaelhalassa.substack.com/p/what-will-schizophrenia-become), I argued that schizophrenia is almost certainly a collection of many entities, and that our diagnostic labels, schizophrenia, schizoaffective disorder, bipolar disorder with psychotic features, carve nature at joints that may not exist or be the best guides for treatment. These categories describe collections of behaviors that may not map on distinct mechanisms or generative processes. From that vantage point, asking whether Nash had “real” schizophrenia or bipolar disorder may be ill posed. The labels are placeholders for something we do not yet understand.
And yet the question refuses to go away, because the clinical differences are real even if the categories are artificial. It is true that I never saw something as dramatic as a beautiful mind, but some people with a schizophrenia diagnosis can have relatively intact cognitive capacity. These people are quite different from those whose cognition appears to deteriorate, akin to Kraepelin’s [dementia praecox](https://en.wikipedia.org/wiki/Dementia_praecox) description. Something is producing that difference, even if our nosology cannot name it. [A paper just published in Molecular Psychiatry by Watson and colleagues](https://www.nature.com/articles/s41380-026-03444-3) takes a step toward naming it, using genetics to do something that clinical observation alone has never managed: begin to show us what is actually varying underneath the label.
[The previous piece I wrote on schizophrenia](https://michaelhalassa.substack.com/p/what-will-schizophrenia-become) argued that the heterogeneity we observe clinically may reflect distinct generative processes rather than variation of a singular category. In other words, among the vast diversity within the [psychosis spectrum](https://pmc.ncbi.nlm.nih.gov/articles/PMC11615062/), we can appreciate at least two distinct trajectories: patients who arrive at their first psychotic break [already cognitively compromised](https://www.nature.com/articles/s41398-023-02718-6), and who face a ceiling on recovery that most cannot exceed, and those whose cognition remains relatively intact, but with the reasoning changes that define psychotic illnesses more generally. If these were simply different severities of one process, you would expect them to respond similarly to treatment and follow predictable paths. They do not.
The question is what to do with that observation. Current clinical tools cannot resolve it. The acute presentation looks the same across both trajectories, and the longitudinal picture only becomes clear years later, often after multiple diagnostic revisions and treatment trials that may not have been appropriate for either. What we need is a way to probe the biology that is upstream of symptoms and independent of the categorical labels that symptoms generate. That is a harder problem than it sounds, and genetics is only one partial answer to it.
Heritability estimates for schizophrenia hover around [~64%](https://pubmed.ncbi.nlm.nih.gov/19150704/) (but removing shared environment, including that in the womb, reduces that further), so genetics alone cannot be the answer and the gap between genetic markers and any clinically actionable prediction remains vast. What genetics appears to be able to do, more modestly, is act as a probe for mechanistic coherence. If a diagnostic category is compressing patients with genuinely distinct underlying biology, the genetic signal inside that category should reflect the compression. It should look composite, pulling in different directions depending on which patients you happen to be sampling. And if you can decompose that signal, you can start to ask whether the clinical heterogeneity you observe maps onto something with a biological basis, even if you cannot yet name the mechanisms.
The cancer parallel is instructive here, even if imperfect. In chronic myelogenous leukemia, a cancer of the blood and bone marrow, decades of clinical heterogeneity (why some patients deteriorated faster, why responses varied) turned out to reflect, in a substantial proportion of cases, the presence of the Philadelphia chromosome. This is a translocation, or swap of genetic material, between chromosomes 9 and 22 that generates an abnormal fusion gene and drives uncontrolled cell growth. Once that mechanism was identified, a drug could be designed against it specifically. Imatinib, [approved in 2001](https://www.nejm.org/doi/full/10.1056/NEJM200104053441401), transformed what had been a death sentence into a manageable condition for many patients. What is critical to understand here is that decomposing a clinically heterogeneous category by its underlying biology changed what was therapeutically possible. Whether that kind of decomposition is achievable in psychosis remains an open question, but Watson and colleagues take a step in that direction, demonstrating that the genetic signal underneath a single diagnostic label is not unitary.
Beginning in the late 2000s, genome-wide association studies (GWAS) made it possible to scan hundreds of thousands of genetic variants across large populations simultaneously, searching for common variants that each contribute a small increment of disease risk. For schizophrenia, the effort eventually identified hundreds of genomic loci. But rather than converging on a coherent biological picture, the signal appeared to pull in multiple directions at once, and understanding why took the better part of a decade.
The first hint of a composite signal came from an apparent paradox within the data itself. At the phenotypic level, schizophrenia is associated with lower educational attainment and cognitive decline. You would therefore expect the genetic risk architecture to show a negative correlation with both. It does not. The genetic correlation between schizophrenia risk and IQ is negative, as expected, but the correlation with educational attainment is weakly positive, meaning the same variants that increase schizophrenia risk also associate, at the population level, with staying in school longer. This counterintuitive finding was reported across multiple large studies and resisted easy explanation.
The resolution, proposed by [Lencz and colleagues](https://www.cell.com/ajhg/fulltext/S0002-9297\(19\)30237-X?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS000292971930237X%3Fshowall%3Dtrue) and subsequently replicated and extended, was that the aggregate GWAS signal was masking two biologically distinct subsets of variants. One subset showed the expected pattern: greater schizophrenia risk alongside lower cognition and lower educational attainment. These variants, when subjected to pathway analysis, pointed toward early neurodevelopmental mechanisms (genes related to how the brain was assembled during early development). A second subset showed the opposite: greater schizophrenia risk alongside higher educational attainment, and pointing towards mechanisms of synaptic function later on in life (mechanisms governing learning after birth). These two components were canceling each other out, resulting in the overall signal being near zero for educational attainment. This was an important conceptual advance, but the methods used could not formally separate and quantify the two components as independent.
A second line of evidence arrived from a different methodology entirely and pointed in a consistent direction. [Rare variant studies](https://www.nature.com/articles/nature07229), which examine low-frequency mutations with large individual effects, found that copy number variants and rare disruptive coding mutations associated with schizophrenia, at loci like 22q11.2, 1q21.1, and 15q13.3, are also enriched in autism and intellectual disability. These are high-penetrance risk factors for neurodevelopmental disruption that happen to increase the probability of psychosis among many other outcomes. Patients carrying heavier rare-variant burden tend to present with [more cognitive compromise](https://www.biologicalpsychiatryjournal.com/article/S0006-3223\(20\)32117-X/abstract).
What the field lacked was a method capable of formally separating the two components of the common-variant signal into distinct genetic constructs, quantifying their independent relationships to cognition, and doing so in a framework clean enough to support downstream analyses like [polygenic scoring](https://www.genome.gov/Health/Genomics-and-Medicine/Polygenic-risk-scores) and [Mendelian randomization](https://en.wikipedia.org/wiki/Mendelian_randomization). That is the gap Watson and colleagues set out to close.
Watson and colleagues took this unresolved picture and asked whether a newer statistical framework could do what prior approaches could not: treat the two components of the schizophrenia signal not as overlapping subsets but as formally independent genetic constructs. The approach they used, GWAS-by-subtraction implemented within a Genomic Structural Equation Modeling (SEM) framework, is worth understanding.
Genomic SEM works similarly in spirit to factor analysis: it models latent genetic dimensions across multiple GWAS datasets simultaneously. The specific application here is intuitive once explained. You start with the full genome-wide association signal for schizophrenia, which captures all common variants associated with the disorder. You then use the bipolar disorder GWAS signal as a genetic filter, statistically removing the variance that schizophrenia shares with affective psychosis. What remains after subtraction is the schizophrenia-specific component: genetic signal that predicts schizophrenia but is independent of affective psychosis risk. The complementary component, the shared portion, is what they call PSYshared.
[](https://substackcdn.com/image/fetch/$s_!7uCD!,f_auto,q_auto:good,fl_progressive:steep/https%3A%2F%2Fsubstack-post-media.s3.amazonaws.com%2Fpublic%2Fimages%2Ff6490a4c-e063-4c5d-a324-015d17e35028_1355x1742.png)
The paper then asks three questions of these components: how do they correlate genetically with IQ and educational attainment (EA), do polygenic scores built from them predict educational outcomes in a large population sample, and does Mendelian randomization support a causal interpretation?
The genetic correlations are where the story sharpens. When you look at the overall schizophrenia signal, it appears to show almost no relationship with educational attainment. That turns out to be because two components are pulling in opposite directions and canceling each other out. The schizophrenia-specific component correlates negatively with both IQ and educational attainment, while the shared component correlates positively with educational attainment.
[](https://substackcdn.com/image/fetch/$s_!tnah!,f_auto,q_auto:good,fl_progressive:steep/https%3A%2F%2Fsubstack-post-media.s3.amazonaws.com%2Fpublic%2Fimages%2Facff7772-5eda-4a9c-b5fa-c1a06eda6af1_1011x686.png)
Polygenic scores built from each component and tested in nearly 382,000 people in the UK Biobank told the same story: people carrying more of the schizophrenia-specific component tended to have lower educational attainment, while those carrying more of the shared component tended to have higher educational attainment. Crucially, this was an entirely independent sample, meaning the opposing signals generalize beyond the original analysis.
Genetic correlations tell you that two things travel together in the genome, but not which way the arrow of causation points, or whether a third factor is driving both. Mendelian randomization addresses this by using genetic variants as a natural experiment. Because you inherit your genes at conception, before any life experiences accumulate, your genetic makeup cannot be caused by your educational outcomes. Watson and colleagues used this approach and found that genetic liability for the schizophrenia-specific component appears to drive lower cognition, while liability for the shared component appears to drive higher educational attainment. The caveat is that some variants may affect both psychosis risk and cognition through completely separate biological routes, so it is difficult to be certain the causal effect is running specifically through psychosis risk rather than through some other property of the same variants. That said, the Mendelian randomization results are broadly consistent with a causal relationship whose veracity will be tested in future experiments.
The final piece of the paper maps where in the brain these two genetic components are most active, using publicly available brain expression databases to ask which regions show the highest expression of the relevant genes. The shared component is predominantly expressed in cortical regions, particularly frontal cortex, consistent with the postnatally expressed synaptic biology that has been associated with affective psychosis in independent work. The schizophrenia-specific component shows a broader pattern, with expression extending into subcortical regions. This pattern is broadly consistent with the picture that has been building across the paper: the schizophrenia-specific component is not simply a more severe version of the shared one, but something with potentially distinct neural involvement. These analyses are exploratory, and the authors present them as such, but they add anatomical texture to the genetic analyses.
So where does this all leave us? Watson and colleagues provide important information about biological heterogeneity within the categorical label of schizophrenia. Maybe more importantly, they clarify the relationship between this category and two others, the psychosis spectrum and neurodevelopmental disorders.
One thing worth noting about the study is the notion of a schizophrenia-specific component. Methodologically, this component is the residual genetic signal that remains after removing shared variance with the broader psychosis spectrum. Personally, I would have probably just called it that, a residual, and not schizophrenia-specific. Because what really is schizophrenia? The neurodevelopmental framing is plausible and supported by converging evidence, but the label risks conferring more categorical definition than the method warrants.
Over many previous pieces in this newsletter, I have made a prediction that what we call schizophrenia will eventually give way to a set of subtypes defined by computational processes altered in individual people. These are algorithmic-level changes that ultimately impact how people build models of the world, update them, and approach reasoning challenges more generally. Two patients carrying the same diagnosis may have distinct algorithmic-level changes that would be described similarly by natural language, the substrate of clinical communication. Do genetics help with this computational dissection? Not directly, but they would add some confidence to conclusions drawn at this downstream process-level description. For grand challenges like schizophrenia, we will need multiple different methods to triangulate the generative process. Genetics underlie vulnerabilities, akin to the hyperparameters that set up model architectures in AI, but all the training data, inputs through early development and later fine-tuning via life, will determine the algorithmic-level changes that shape clinical presentation, and perhaps even treatment response.
So did John Nash really have schizophrenia? The film that brought his story to millions took considerable liberties with the clinical record: his hallucinations were auditory, not the elaborate visual conspiracies Hollywood rendered; and his greatest mathematical work was complete before psychosis ever emerged. There had been [some discussion](https://onlinelibrary.wiley.com/doi/10.1111/acps.12504) regarding whether his condition was truly schizophrenia or bipolar, but that debate may miss the point entirely. What's clear from the record is that Nash was not a man who did mathematics through schizophrenia, he was someone whose psychosis arrived after his most important work was done, perhaps ran an episodic illness and eventually receded, leaving behind someone who was diminished in some ways but still capable of serious thought. That trajectory does not sit outside the distribution of what we call schizophrenia. **If individual people carry the neurodevelopmental and affective psychosis components in continuously varying proportions, then Nash's case stops being a paradox and becomes exactly what we should expect to find somewhere in that space.** His case has been sitting at the edge of our nosology for decades, neither fully explained nor fully dismissed. Now we have tools to reframe the question entirely.
[](https://substackcdn.com/image/fetch/$s_!PQfr!,f_auto,q_auto:good,fl_progressive:steep/https%3A%2F%2Fsubstack-post-media.s3.amazonaws.com%2Fpublic%2Fimages%2F881341fc-fc7c-4c5f-a7fc-bc73d4cda327_640x524.gif)
*If you found this piece useful, please consider subscribing and sharing it with someone who might appreciate it too.*
Acknowledgement: I am grateful for Dr. [Cameron Watson](https://www.kcl.ac.uk/people/cameron-watson)’s input on this post. Please check him out and follow his work. I know I will\!
[](https://substackcdn.com/image/fetch/$s_!29CN!,f_auto,q_auto:good,fl_progressive:steep/https%3A%2F%2Fsubstack-post-media.s3.amazonaws.com%2Fpublic%2Fimages%2F684d07dd-50b9-4715-b5b8-cc9f6d1db584_200x200.png)
Cameron is a Specialist Registrar in Psychiatry at the Maudsley Hospital in London and a UKRI Medical Research Council Clinical Research Training Fellow at the Institute of Psychiatry, Psychology and Neuroscience, King’s College London. His research integrates cognitive, genetic, and neurophysiological data to disentangle heterogeneity within psychotic disorders, with the ultimate aim of improving understanding of the diverse mechanisms driving these conditions.
No posts |
| Shard | 76 (laksa) |
| Root Hash | 14862242593741677076 |
| Unparsed URL | com,substack!michaelhalassa,/p/did-john-nash-really-have-schizophrenia s443 |